A Retinite Pigmentosa (RP), também conhecida como Retinose Pigmentar, é o termo utilizado para designar uma doença degenerativa da retina que causa perda de visão progressiva, podendo levar à cegueira. Esta condição se manifesta quando as células da retina, chamadas fotorreceptores, não funcionam como deveriam.
A retinite pigmentosa é uma desordem rara e hereditária, que causa degeneração na retina, responsável por capturar as imagens a partir do campo visual. A retinite afeta os fotorreceptores bastonetes e cones, que são as células sensíveis à luz e responsáveis pela visão periférica e noturna.
Por causa dessa condição, as células começam a se degenerar, atrofiam e morrem. A retinite geralmente se manifesta em jovens adultos e adolescentes, mas pode também ser diagnosticada na infância, como a amaurose congênita de Leber, que é a forma precoce e grave da doença.
A retinite pigmentosa geralmente começa na infância, mas a idade aproximada que a condição surge e a rapidez com que se desenvolve varia de pessoa para pessoa. A maioria das pessoas com essa condição perde grande parte da visão no início da idade adulta.
Como os bastonetes geralmente são afetados primeiro, o primeiro sintoma que se pode notar é que leva mais tempo para a visão se ajustar à escuridão (chamada de “cegueira noturna“). Por exemplo, passar a tropeçar em objetos no escuro ou não conseguir dirigir à noite.
Quem possui essa condição pode perder a visão periférica ao mesmo tempo ou logo após a visão noturna diminuir. É possível também apresentar a “visão de túnel”, que é a perda da capacidade de enxergar sem precisar virar a cabeça para o lado, ou seja, a visão periférica é prejudicada.
Nos estágios posteriores, os cones podem ser afetados. Isso tornará mais difícil realizar trabalhos detalhados e ainda pode ocasionar na dificuldade em ver cores. É raro, mas às vezes os cones são os primeiros a se degenerarem.
Em alguns casos, há também a presença de fotofobia, condição em que a pessoa sente desconforto em ambientes com fortes luzes, ou da fotopsia, onde a visão é afetada por contínuos flashes de luz que brilham e piscam.
Mais de 60 genes diferentes podem causar os diferentes tipos de retinite pigmentosa. Os pais podem transmitir os genes problemáticos aos seus filhos de três maneiras diferentes:
Retinite pigmentosa autossômica recessiva: cada pai tem uma cópia da condição e uma cópia normal do gene responsável, mas não apresenta nenhum sintoma. Uma criança que herda duas cópias problemáticas do gene (uma de cada pai) desenvolverá este tipo de retinite pigmentosa. Como são necessárias duas cópias do gene problemático, cada criança da família tem 25% de chance de ser afetada.
Retinite pigmentosa autossômica dominante: esse tipo de retinite pigmentosa requer apenas uma cópia do gene problemático a ser desenvolvido. Um pai com esse gene tem 50% de chance de passá-lo para cada criança.
Retinite pigmentosa ligada ao X: Uma mãe que carrega o gene problemático pode transmiti-lo para seus filhos. Cada um deles tem 50% de chance de desenvolver a condição. A maioria das mulheres portadoras do gene não apresenta sintomas, mas cerca de 1 em cada 5 terá sintomas leves.
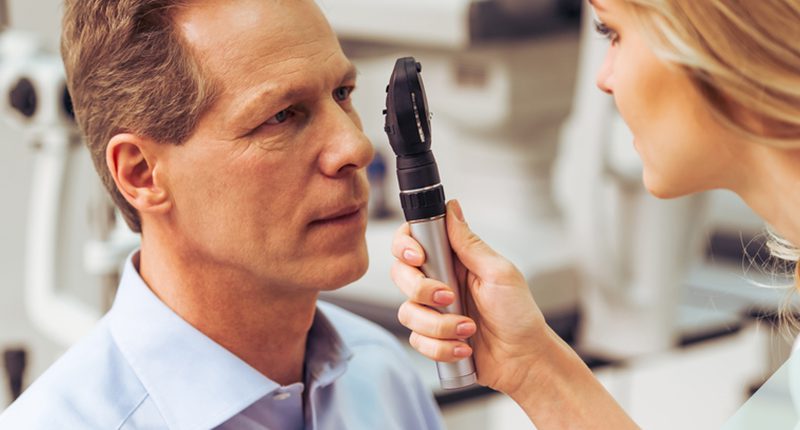
Um oftalmologista pode dizer se você tem retinite pigmentosa. Ele pode fazer uma avaliação através dos seguintes exames oftalmológicos:
Fundoscopia: O médico utiliza um colírio para dilatar a pupila e obter uma melhor visão da retina. Esse exame permite que ele enxergue a parte do fundo do olho e avalie o nervo óptico, a retina, os vasos e a mácula. Se o paciente tiver retinite, haverá tipos específicos de manchas escuras na retina;
Teste de campo visual: é utilizado para avaliar se o campo de visão central e periférico estão normais ou se existem alterações em algum ponto da visão;
Eletrorretinograma: registra os potenciais elétricos da retina através de eletrodos, sob forma de lente de contato, colocados “sobre” a córnea.
Se você ou alguém da sua família for diagnosticado com retinite pigmentosa, todos os membros da família devem procurar o oftalmologista para fazer o diagnóstico.
Ainda não há cura para a retinite pigmentosa, mas os cientistas estão trabalhando para encontrar novos tratamentos. Algumas opções podem retardar sua perda de visão e até mesmo restaurar a visão já perdida em decorrência dessa condição:
Acetazolamida: Nos estágios finais, a pequena área no centro da retina pode inchar. Isso é chamado de edema macular e também pode reduzir a visão. Este medicamento pode aliviar o inchaço e melhorar a visão.
Palmitato de vitamina A: altas doses deste composto podem retardar um pouco a retinite pigmentosa.
Uso de óculos de sol: Estes tornam os olhos menos sensíveis à luz e os protegem dos raios ultravioleta prejudiciais que podem acelerar a perda de visão.
Implante de Retina: Um cirurgião coloca um dispositivo eletrônico dentro e ao redor dos olhos. Quando combinada com óculos especiais, permite que algumas pessoas com retinite pigmentosa em estágio avançado voltem a ler, mesmo que somente letras em fonte maiores, além de se movimentarem sem uma bengala ou cão-guia.

Junte-se a nossa lista para receber dicas sobre oftalmologia por e-mail
Junte-se a nossa lista para receber dicas sobre oftalmologia por e-mail